












































































What’s Next in the Evolution of Standards for Biologics Development


Chief Science Officer
United States Pharmacopeia (USP)
Documentary standards such as monographs, general chapters, and reference standards are essential for efficiently meeting regulatory requirements and ensuring the intended efficacy and safety of drugs. At the most fundamental level, compendial standards for medicines, set by pharmacopeias such as the United States Pharmacopeia (USP), address common requirements such as sterility, endotoxin limits, particulate matter, and microbial contamination.
At the product level, standards can address the measurement of specific quality attributes for different therapeutic modalities. For small-molecule drugs, the use of standards plays an important role in promoting competition and reducing medicine costs1.
For biologics, standards are needed more than ever as novel modalities become a cornerstone of healthcare.2 Monoclonal antibodies (mAbs) and their biosimilars (follow-on versions of approved biologics) are among the most widely used biologics today. Since the first mAb was approved by the U.S. Food and Drug Administration (FDA) in 1986, more than 100 therapeutic mAb drugs are now on the market. In recent years, novel antibody formats such as antibody-drug conjugates (ADCs), bispecific antibodies, fusion proteins, and other biologics have shown significant growth.
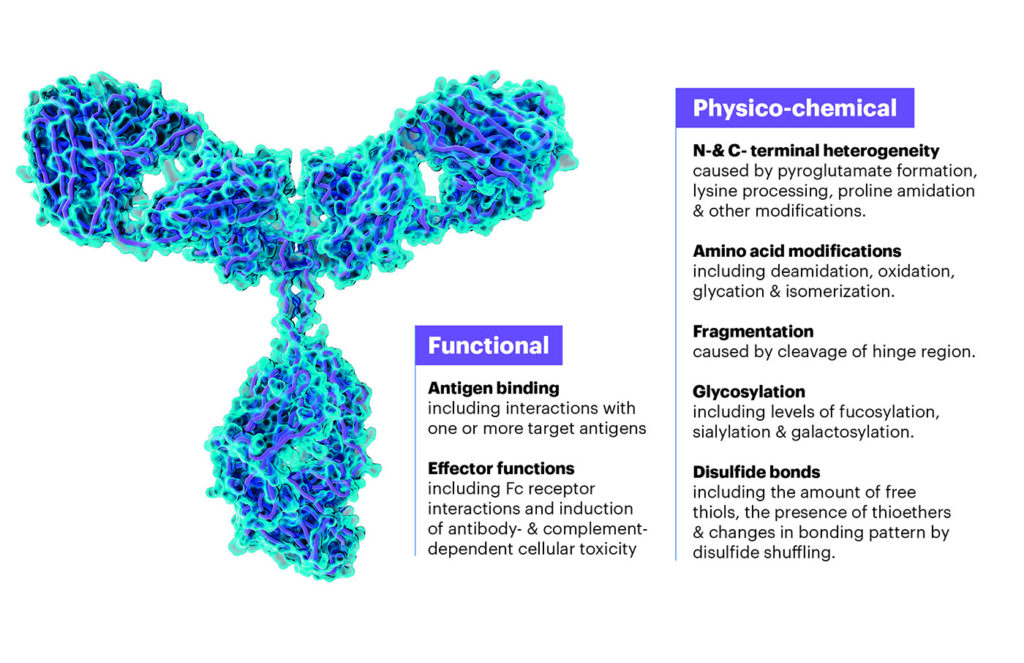
[Adapted from Atouf and Venema.3 usp]
Why biological standards matter
Biological standards play a critical role in enabling innovation, ensuring product quality, and protecting patient safety across the product lifecycle. By providing established methods and reference materials, standards eliminate the costly and time-consuming duplication of effort required for manufacturers to develop and validate their own approaches to assessing product quality.
For biosimilar developers, access to globally harmonized standards can enable access to methods vetted through a public standard-setting process and subsequently support a competitive marketplace. Standards also provide a stable foundation as analytical technologies evolve, allowing legacy methods to be bridged with newer techniques while maintaining confidence in data continuity and comparability over time.
For therapeutic proteins, including mAbs, standards help define and control critical quality attributes. These attributes influence bioavailability, clearance, and functional activity and must be managed through robust control strategies that minimize manufacturing variability. By establishing common benchmarks and measurement tools, standards enable consistency.
Harmonized standards help ensure manufacturing and lot-to-lot consistency and support reliable comparison of products over time, giving patients confidence that efficacy and safety are maintained when switching between an innovator product and a biosimilar or among biosimilars.
From a regulatory perspective, standards create a common foundation for streamlined evaluation. When products’ dossiers are submitted using compendial methods, regulatory agencies such as the FDA and European Medicines Agency (EMA) traditionally accept these methods to demonstrate compliance with certain quality attributes, enabling more predictable regulatory review while preserving regulatory authority over final determinations of equivalence.
The evolution of biologics standards
The evolution of standards for biologics reflects the growing complexity and maturity of the field, highlighting the need for both general and product-specific guidance. Early efforts by pharmacopeias mirrored the approach used for small molecules, creating individual monographs for each biologic drug. This approach worked for simpler biologics like insulin or growth hormone, which could be reliably deployed using a defined set of standardized tests.
However, as biologics became more complex, this model became increasingly challenging. Variability inherent to these molecules, often due to differences in manufacturing processes, made it difficult to define a single set of acceptance criteria that could consistently apply across products.
In response, in 2010, our focus shifted toward developing standards that apply across product classes to support the analysis of product quality attributes (PQAs). These included broad, platform-based methods for measuring characteristics such as glycosylation, aggregation, and residual DNA, attributes shared across product classes or expression systems.
These general chapters provided a strong foundational framework, which can be used throughout the product lifecycle and across diverse biologic products. There is now growing recognition of the complementary need for product-specific standards, especially for therapeutic proteins, which make up a significant portion of the biologics market.
While general standards address common analytical needs, individual biologics like trastuzumab or erythropoietin have unique structural and functional characteristics that benefit from more tailored methods. Rather than shifting from one type of standard to another, the most effective approach blends both, supporting innovation while ensuring consistency, comparability, and regulatory confidence throughout the product lifecycle.
To support ensuring the quality of complex biological products, these approaches are used:
- Standards for individual biologic drugs, which historically focused on legacy biologics such as insulin drugs
- Standards for cross-cutting attributes (e.g., measurement of glycosylation across biologics)
- Platform-based methods and standards for testing products sharing production systems (e.g., mAbs made in CHO cells)
USP is now engaging stakeholders to explore proposals for flexible documentary standards to support the quality of drug substances used in a wide range of biological products. These standards support biologics, including biosimilars, in three ways:
- Ensuring consistency when making process changes or introducing new manufacturing processes
- Enabling comparative analytical assessment when products move from single to multi-manufacturer settings
- Detecting substandard products, which can otherwise go unnoticed without an appropriate benchmark
Evolving biologics and standards
As biologic products have evolved, they have become increasingly complex, expanding beyond mAbs to include ADCs, cell and gene therapies (CGTs), genome-editing platforms, and mRNA-based products. Standards remain critical to ensuring product quality, consistency, and patient safety, but those for emerging modalities cannot always take the same form as those applied to small molecules or conventional biologics.
Next-generation therapies introduce new quality challenges that extend beyond the final drug product to individual components and upstream processes. For example, CRISPR-based therapies combine guide RNA, enzymes, and DNA templates, making it essential to control and characterize the quality of each building block. This represents a shift from a traditional end-product focus to a more integrated approach in which quality must be established throughout development and manufacturing.
At the same time, not all advanced therapies are well-suited to traditional product-specific monographs. Some CGTs are highly individualized, developed for a single patient or small populations, making classical monographs impractical or unnecessary. In these cases, standards that address shared critical quality attributes across a product class or manufacturing platform are more appropriate.
As scientific understanding and manufacturing practices mature, opportunities may emerge to develop more targeted standards that address challenges associated with scaling and consistency of manufacturing processes.
mRNA-based therapies further illustrate this evolving landscape. While mRNA has a defined nucleotide sequence that could eventually enable biosimilar development, standards must address not only physicochemical attributes such as sequence integrity and purity, but also functional performance that varies depending on intended use, whether vaccination or therapeutic protein expression. Cross-cutting standards that support common analytical needs therefore represent a critical starting point, with more tailored standards following as consensus develops.
Recognizing these realities, we are proposing a hybrid model designed to balance scientific rigor with flexibility. While monographs remain the gold standard for defining identity, strength, purity, and quality of small molecules and some well-characterized biologics, advanced modalities often require alternative approaches. The framework includes:
- Informational general chapters that provide best practices and shared scientific understanding across product classes
- Emerging standards that introduce early developmental concepts and invite community engagement
- Procedural general chapters with accompanying reference standards to assess quality attributes common across a product class
- Analytical Reference Materials (ARMs) that provide early benchmarks for assay development. These may evolve into reference standards to establish system suitability and to monitor assay performance for physicochemical and functional CQAs.
Over time, elements of this hybrid approach may mature into compendial general chapters or full monographs, for example, when an emerging method for mRNA purity gains sufficient regulatory and industry support. This model maintains public trust and scientific rigor while remaining responsive to innovation and enabling global harmonization.
Importantly, as advanced therapies mature and move into broader clinical and commercial use, the need for product-specific standards becomes increasingly important. Platform and cross-cutting standards establish a critical foundation, but they cannot fully capture the unique attributes and risks associated with individual products. Product-specific standards provide the precision needed to guide development, support comparability, and establish robust control strategies, accelerating progress while reducing duplication, cost, and uncertainty.
Meaningful standards are only valuable if they are developed using scientific and public processes, adopted, and implemented. We must equip scientists and manufacturers with the knowledge to apply those tools effectively and cultivate awareness across the broader community about the value of standards.
Continued collaboration among regulators, industry leaders, and standards-setting organizations is critical. Together, we can shape a standards ecosystem that keeps pace with scientific advances while providing the clarity and consistency needed for global development and regulatory approval, patient access, and patient health.
References
1. Murimi-Worstell B, Ballreich M, Seamans G, Alexander, C. Association between US Pharmacopeia (USP) monograph standards, generic entry and prescription drug costs. Published: November 12, 2019.
2. Gupta, RK. The vital role of biological standardization in ensuring efficacy and safety of biological products–Historical perspectives. Journal of Pharmaceutical Sciences, 2025; 114(2): 690–700.
3. Atouf F and Venema J. Do Standards Matter? What is Their Value? Journal of Pharmaceutical Sciences, 2020; 109(8): 2387-2392.
Fouad Atouf, PhD, is the chief science officer at United States Pharmacopeia.
The post What’s Next in the Evolution of Standards for Biologics Development appeared first on GEN - Genetic Engineering and Biotechnology News.
Apa Reaksi Anda?
 Suka
0
Suka
0
 Kurang Suka
0
Kurang Suka
0
 Setuju
0
Setuju
0
 Tidak Setuju
0
Tidak Setuju
0
 Bagus
0
Bagus
0
 Berguna
0
Berguna
0
 Hebat
0
Hebat
0











