








































































A New Development Playbook for PROTACs

International Marketing Associate Director, La, boratory Testing Division, WuXi AppTec
As proteolysis-targeting chimeras (PROTACs) mature from scientific breakthrough to clinical modality, a candidate’s degradation potency is no longer enough to justify its advancement. A strong degrader is not necessarily a strong drug candidate if liabilities in exposure, safety, selectivity, manufacturability, or dosing strategy emerge later. As the field advances, success will depend on whether sponsors can integrate chemistry, pharmacology, safety, manufacturability, and clinical strategy early enough to translate promising degraders into viable medicines.
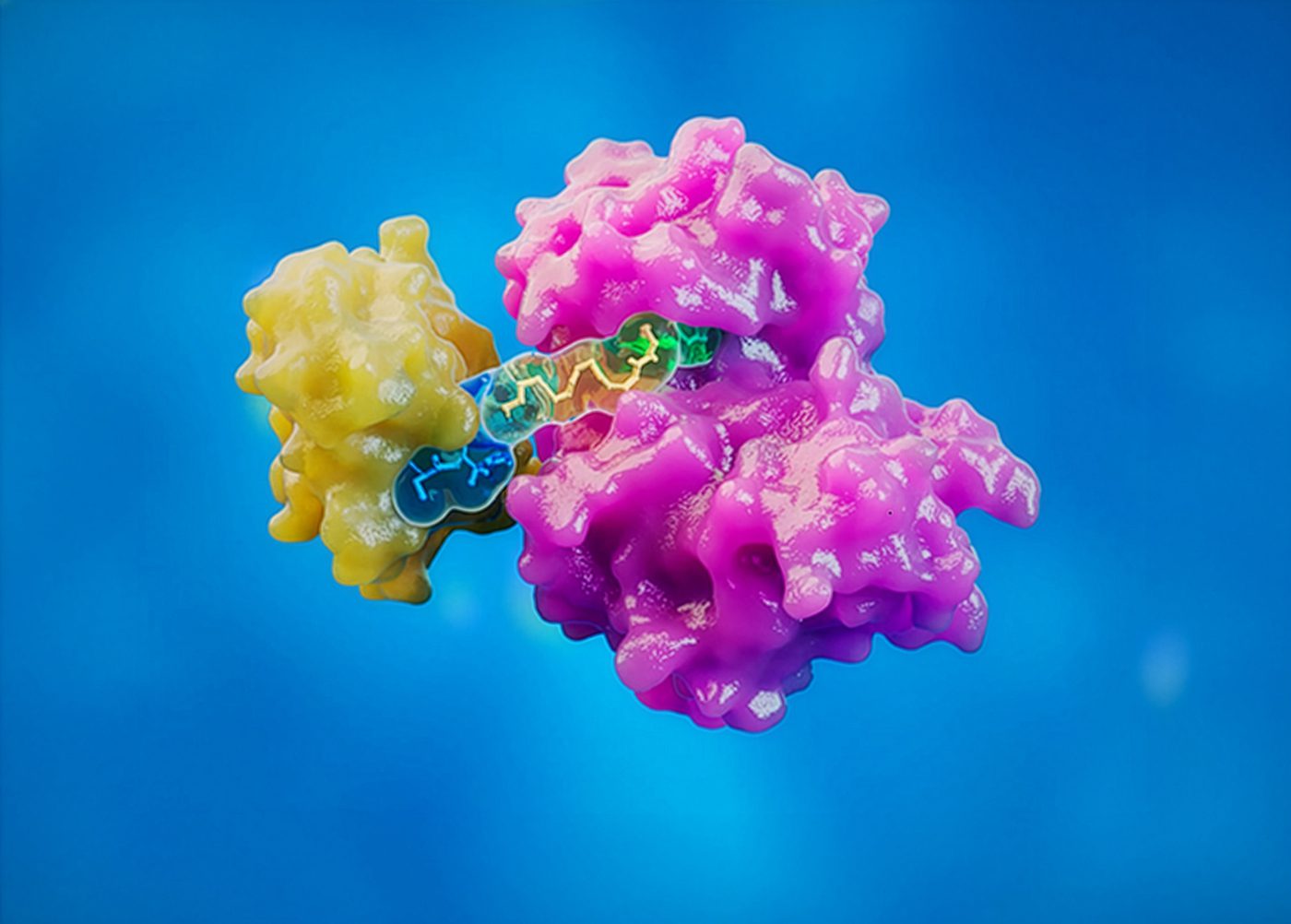
PROTACs have helped redefine what may be possible in drug discovery. By harnessing the ubiquitin-proteasome system to eliminate disease-relevant proteins, rather than simply inhibiting their activity, they have expanded the scope of targets that may be therapeutically addressable. For the past several years, much of the excitement around PROTACs has centered on this breakthrough mechanism. The field has been driven by the promise of degrading previously “undruggable” proteins, improving selectivity, and potentially overcoming resistance mechanisms that limit traditional inhibitors. But as clinical programs progress and the modality moves closer to late-stage regulatory milestones, the central question is changing.
The issue is no longer whether targeted protein degradation works. It is whether promising degraders can be translated into clinically and commercially viable therapies.
That transition marks an important phase of maturity for PROTAC development. The next wave of progress will depend on translational discipline and the ability to balance potency with developability, pharmacology, safety, manufacturability, and clinical feasibility from the earliest stages of development.
Entering a new phase of maturity
Early PROTAC innovation was rightly focused on validating the modality itself. Demonstrating that a heterobifunctional molecule could recruit an E3 ligase, drive ubiquitination of a target protein, and induce selective degradation was a foundational scientific achievement. That early work established targeted protein degradation as part of a broader wave of transformational therapeutic modalities, alongside approaches such as RNA interference therapeutics and antibody–drug conjugates, that have expanded what drug developers can target and how they think about translation.
Today, however, the field is operating under a different set of expectations. As more candidates advance through clinical development, sponsors must show not just that a PROTAC can degrade a target, but that it can do so with an exposure profile, safety margin, formulation strategy, and manufacturing pathway appropriate for clinical applications. A potent degrader in vitro may still fail to become a viable development candidate if it cannot achieve sufficient intracellular exposure, is metabolically unstable, if its degradation profile extends beyond the intended target set, or if its chemistry introduces manufacturing and formulation complications that slow advancement.
In other words, the scientific novelty of a modality can carry a program only so far before technical feasibility must be addressed for a candidate to advance. For PROTACs, that moment has arrived.
Potency alone is an incomplete metric
Degradation potency remains important. Maximum degradation, degradation half-life, and related pharmacodynamic measures are essential for understanding whether a molecule is engaging its biology as intended. But potency on its own can be misleading, particularly when it becomes the dominant criterion for candidate selection.
PROTACs are not conventional inhibitors. Their event-driven, catalytic mechanism introduces complexities that make exposure-response relationships less intuitive than those seen with traditional small molecules. Biological effects may persist after plasma concentrations decline, while higher concentrations do not necessarily lead to greater activity. In some cases, excessive exposure may even reduce degradation efficiency because of saturation effects that limit productive ternary complex formation.
This means the “best degrader” in a screening cascade is not always the best drug candidate. A molecule may demonstrate impressive degradation in a cellular assay while carrying liabilities that emerge only later, such as poor permeability, limited oral bioavailability, rapid linker metabolism, high nonspecific binding, unstable analytical performance, or off-target degradation driven by ligase biology or ternary complex behavior. If those issues are not considered early, potency can create a false sense of confidence in a degrader’s potential for clinical use.
Development workflows fall short
One reason translational issues emerge so frequently in PROTAC programs is that many development workflows still reflect assumptions built around traditional small molecules. In those models, discovery, DMPK, bioanalysis, toxicology, and chemistry, manufacturing, and controls (CMC) often proceed in a staged or partially sequential manner, with each function evaluating modality-relevant properties within its own domain before handing it forward.
With targeted protein degraders, however, early chemistry decisions can directly influence permeability, intracellular exposure, metabolic clearance, assay reliability, biodistribution, and manufacturability. Linker design, ligand selection, and overall polarity are not simply medicinal chemistry concerns; they shape how the molecule behaves across the entire development continuum. Likewise, a bioanalytical challenge may obscure the interpretation of PK/PD relationships, complicate dose optimization, or delay confidence in candidate selection.
The same is true for safety. Because PROTACs eliminate proteins rather than transiently inhibiting them, the consequences of target engagement can differ meaningfully from those associated with conventional inhibitors. On-target toxicity may emerge when complete or prolonged degradation is not tolerated, even if partial functional inhibition is acceptable. Off-target effects may arise not only from target promiscuity, but also from E3 ligase recruitment and unintended ternary complex formation. These risks cannot be addressed effectively if safety is considered only after potency and exposure have been optimized.
Traditional workflows can also underweight manufacturability and CMC considerations. PROTACs are generally handled as small molecules, but their structural complexity can create multi-step synthesis challenges, impurity-control difficulties, and formulation constraints much earlier than teams may expect. When these issues are discovered late, promising programs can lose momentum for reasons that have little to do with biology.
The core issue is not organizational design alone. It is that PROTACs expose the limits of linear decision-making. They require earlier integration because the liabilities that determine success are tightly interconnected.
PROTAC-specific development
If PROTACs require a different development model, what would it look like?
First, a successful PROTAC development plan should begin with balanced optimization across parameters rather than sequential, single-parameter optimization. Candidate selection should account not only for degradation potency, but also for permeability, solubility, metabolic stability, intracellular exposure, selectivity, formulation feasibility, and synthetic tractability. Programs that rank candidates holistically are better positioned to recognize which molecules are genuinely translatable.
Second, the PK/PD strategy should be built around the biology of degradation. Because systemic exposure does not fully explain pharmacological effect, teams increasingly need direct measures of target degradation and recovery kinetics, not just plasma concentration data. Mechanistic PK/PD models can help connect degradation durability, protein resynthesis, and dosing schedule in a way that better reflects how PROTACs work in vivo.
Third, bioanalysis should be treated as a strategic enabler rather than a downstream technical function. PROTACs can introduce assay complications, including nonspecific binding, chromatographic artifacts, and instability across matrices. Robust analytical methods are essential not only for quantitation but for making reliable decisions about exposure, disposition, and translation across study systems.
Fourth, safety assessment must expand beyond conventional assumptions. Early proteomic profiling, tissue distribution analysis, and evaluation of degradation selectivity can help identify liabilities before they become entrenched in a program. For PROTACs, understanding where degradation occurs, how long it persists, and what unintended proteins may be affected is central to designing an acceptable therapeutic window.
Finally, CMC and manufacturability should be considered earlier than many teams may be accustomed to. A molecule with compelling pharmacology but limited synthetic scalability, poor solid-state properties, or unstable formulation behavior may not be a strong development candidate. Integrating these realities earlier supports smarter program prioritization and reduces late-stage surprises.
Taken together, these elements define a development playbook centered on translation. They signal a maturation of the field, in which the emphasis shifts from demonstrating biological power to establishing overall developability, recognizing that promising degraders must ultimately succeed as integrated therapeutic candidates, not just mechanistic innovations.
Sponsors can improve the odds
For sponsors advancing PROTAC programs, translation should be a design principle from the beginning. That starts with cross-functional alignment early in discovery. Chemistry, DMPK, bioanalysis, safety, and CMC teams should work together to shape candidate criteria, so that trade-offs are recognized early, and optimization reflects the realities of development rather than the priorities of any one function.
It also means adopting more realistic success metrics. Degradation data should remain central, but it should be interpreted alongside developability, not in isolation from it. Sponsors may also benefit from building translational assays and biomarkers earlier. The ability to directly measure target degradation and connect it to pharmacodynamic effect can strengthen decision-making throughout preclinical and clinical development. In a modality where traditional exposure markers may be incomplete, translational pharmacology can provide strategic direction.
Most importantly, teams should resist the temptation to force PROTACs into a conventional small-molecule framework. These candidates may be classified as small molecules for many regulatory purposes, but functionally, they behave as a distinct modality. Treating them as such allows the development strategy to evolve in step with biology.
The next phase of PROTAC success
PROTACs have already shifted the pharmacological landscape around drugability. Their next contribution may be just as important by forcing the industry to rethink what a good development strategy looks like for complex, mechanism-driven therapeutics. As the field matures, successful drug sponsors will be those who can translate degradation into a developable, manufacturable, safe, and clinically meaningful therapy. That requires a different playbook built on integration, balanced optimization, and translational discipline. For PROTACs, that is no longer a future concern. It is the central challenge of the present.
Shanghao Li, PhD, currently serves as international marketing associate director in the Laboratory Testing Division at WuXi AppTec.
The post A New Development Playbook for PROTACs appeared first on GEN - Genetic Engineering and Biotechnology News.
Apa Reaksi Anda?
 Suka
0
Suka
0
 Kurang Suka
0
Kurang Suka
0
 Setuju
0
Setuju
0
 Tidak Setuju
0
Tidak Setuju
0
 Bagus
0
Bagus
0
 Berguna
0
Berguna
0
 Hebat
0
Hebat
0